
1. Ethorphine
2. Etorphine
3. M99
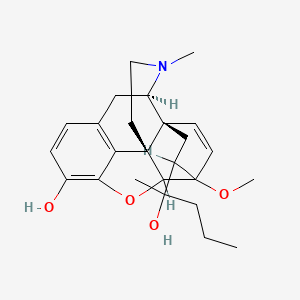
1. Etorphine
2. 14521-96-1
3. (1r,2s,6r)-19-(2-hydroxypentan-2-yl)-15-methoxy-5-methyl-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11,16-tetraen-11-ol
4. (1r,2s,6r,14r,15r,19r)-19-[(2s)-2-hydroxypentan-2-yl]-15-methoxy-5-methyl-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11,16-tetraen-11-ol
5. Pdsp2_001571
6. Db01497
7. Q416827
| Molecular Weight | 411.5 g/mol |
|---|---|
| Molecular Formula | C25H33NO4 |
| XLogP3 | 3.1 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 4 |
| Exact Mass | 411.24095853 g/mol |
| Monoisotopic Mass | 411.24095853 g/mol |
| Topological Polar Surface Area | 62.2 Ų |
| Heavy Atom Count | 30 |
| Formal Charge | 0 |
| Complexity | 772 |
| Isotope Atom Count | 0 |
| Defined Atom Stereocenter Count | 3 |
| Undefined Atom Stereocenter Count | 4 |
| Defined Bond Stereocenter Count | 0 |
| Undefined Bond Stereocenter Count | 0 |
| Covalently Bonded Unit Count | 1 |
/VET/: Thirty-five anesthetic events involving 15 captive addax (Addax nasonzaculatus) were performed between August 1998 and February 2002 using a combination of etorphine (33.7 +/- 7.9ug/kg) and detomidine (21.9 +/- 4.6 ug/ kg) or a combination of medetomidine (57.4 +/- 8.6 ug/kg) and ketamine (1.22 +/- 0.3 Ug/kg), with or without supplemental injectable or inhalant anesthetic agents. Etorphine-detomidine anesthesia was antagonized with diprenorphine (107.1 +/- 16.4 ug/kg) and atipamezole (100.9 +/- 42.4 ug/kg). Medetomidine-ketamine anesthesia was antagonized with atipamezole (245.3 +/- 63.4 ug/kg). Animals became recumbent within 5 min when the combination of etorphine and detomidine was used and within 11 min when the combination of medetomidine and ketamine was used. Both drug combinations were suitable for use as primary immobilizing agents producing short-duration restraint and analgesia. Bradycardia was noted with both combinations. Further investigation of the cardiopulmonary effects of both combinations is warranted.
PMID:14582789 Portas TJ et al; J Zoo Wildl Med 34 (3): 269-73 (2003)
/VET/: Implantation or injectable dosage form new animal drugs. Etorphine hydrochloride injection. ... Conditions of use. (1) The drug is used for the immobilization of wild and exotic animals. (2) It is administered intramuscularly by hand syringe or syringe dart at a suitable dosage level depending upon the species. (3) Do not use the drug unless diprenorphine hydrochloride injection, veterinary, as provided for in section 522.723, is available for use in reversing the effects of etorphine hydrochloride injection, veterinary. (4) For use in wild or exotic animals only. Do not use in domestic food-producing animals. Do not use 30 days before, or during, the hunting season in free-ranging wild animals that might be used for food. (5) Federal law restricts this drug to use by or on the order of a licensed veterinarian. Distribution is restricted to veterinarians engaged in zoo and exotic animal practice, wildlife management programs, and researchers. /Etorphine Hydrochloride/
21 CFR 522.883 (USFDA); U.S. National Archives and Records Administration's Electronic Code of Federal Regulations. Available from, as of March 24, 2008: https://www.ecfr.gov
/VET/: Captive rhinoceros species are most frequently sedated and/or anesthetized with the potent opioid, etorphine hydrochloride in combination with an alpha-2 adrenoreceptor agonist or the butyrophenone, azaperone. Carfentanil citrate based combinations have also been used to a lesser extent. In recent years butorphanol tartrate based combinations have been used with good success to induce neuroleptanalgesia. Sedation and anesthesia are complicated by the large size of all rhinoceros species and their sensitivity to potent opioids. Potential complications include respiratory depression, hypoxaemia, hypertension, pulmonary shunting and ventilation/perfusion mismatch.
PMID:15478725 Portas TJ; Aust Vet J 82 (9): 542-9 (2004)
/VET/: Use with extreme care: very small amounts may bring about respiratory paralysis and death.
O'Neil, M.J. (ed.). The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals. Whitehouse Station, NJ: Merck and Co., Inc., 2006., p. 663
Etorphine is only available for its use in veterinary. This main usage is related to the immobilization of large mammals.
Etorphine is a synthetic cousin of morphine and 40,000 times more powerful.
Hypnotics and Sedatives
Drugs used to induce drowsiness or sleep or to reduce psychological excitement or anxiety. (See all compounds classified as Hypnotics and Sedatives.)
Narcotics
Agents that induce NARCOSIS. Narcotics include agents that cause somnolence or induced sleep (STUPOR); natural or synthetic derivatives of OPIUM or MORPHINE or any substance that has such effects. They are potent inducers of ANALGESIA and OPIOID-RELATED DISORDERS. (See all compounds classified as Narcotics.)
Analgesics, Opioid
Compounds with activity like OPIATE ALKALOIDS, acting at OPIOID RECEPTORS. Properties include induction of ANALGESIA or NARCOSIS. (See all compounds classified as Analgesics, Opioid.)
Eleven juvenile African elephants were given etorphine hydrochloride (2.19 +/- 0.11 micrograms/kg of body weight; mean +/- SD) as a single IM injection; 3 elephants were given additional etorphine (0.42 +/- 0.09 micrograms/kg) IV. After immobilization, each elephant was maintained in lateral recumbency by administration of a 0.5% halothane/oxygen mixture or by administration of multiple IV injections of etorphine. At postinjection hours 0.25 and 0.5 and at 30-minute intervals thereafter, blood samples were collected via an auricular artery, and serum concentrations of etorphine were determined by use of radioimmunoassay. The highest mean serum concentration of etorphine in 6 elephants given a single IM injection and subsequently maintained on halothane and oxygen was 1.62 +/- 0.97 ng/mL at postinjection hours 0.5; thereafter, the mean serum concentration decreased steadily. In 4 elephants maintained in lateral recumbency with multiple IV administrations of etorphine, a correlation was not found between the time to develop initial signs of arousal and serum concentrations of etorphine before arousal. After administration of the initial immobilizing dose of etorphine, the interval between successive IV administrations of etorphine decreased. /Etorphine hydrochloride/
PMID:3505931 Jacobson ER et al; J Am Vet Med Assoc 189 (9): 1079-81(1986)
In a forensic case involving Immobilon /commerical formulation/, the etorphine concentrations measured in postmortem femoral vein and heart blood specimens were 14.5 and 23.5 ug/L, respectively. No etorphine was detected in the urine.
PMID:10376332 Elliott SP, Hale KA; Forensic Sci Int 101 (1):9-16 (1999)
Etorphine is an agonist at mu, delta, and kappa opioid receptors. It also has a weak affinity for the ORL1 nociceptin/orphanin FQ receptor.
A growing body of evidences suggests that receptor desensitization is implicated in the development of tolerance to opioids, which is generally regulated by protein kinases and receptor trafficking proteins. In the present study, we demonstrated that repeated s.c. treatment with etorphine, but not morphine, produced a significant increase in protein levels of G protein-coupled receptor kinase 2, dynamin II, beta-arrestin 2 and phosphorylated-conventional protein kinase C in membranes of the mouse spinal cord, suggesting that the etorphine-induced mu-opioid receptor desensitization may result from G protein-coupled receptor kinase 2/dynaminII/beta-arrestin2-dependent phosphorylation of mu-opioid receptors. Unlike etorphine, morphine failed to change the levels of these trafficking proteins. Furthermore, we found that the level of glial fibrillary acidic protein in the mouse spinal cord was clearly increased by chronic in vivo and in vitro treatment with morphine, whereas no such effect was noted by etorphine. In the behavioral study, intraperitoneal pretreatment with the glial-modulating agent propentofylline suppressed the development of tolerance to morphine-induced antinociception. In addition, intrathecal injection of astrocytes and astrocyte-conditioned medium mixture, which were obtained from cultured astrocytes of the newborn mouse spinal cord, aggravated the development of tolerance to morphine. In contrast, these agents failed to affect the development of tolerance induced by etorphine. These findings provide direct evidence for the distinct mechanisms between etorphine and morphine on the development of tolerance to spinal antinociception. These findings raise the possibility that the increased astroglia response produced by chronic morphine could be associated with the lack of mu-opioid receptor internalization.
PMID:16417975 Narita M et al; Neuroscience 138 (2): 609-19 (2006)
The cellular location of extracellular signal-regulated kinases (ERKs) activated by a G protein-coupled receptor was shown to be dependent on the pathway that mediated their activation. In general, fast activation of ERKs (2 min) mediated by G proteins resulted in the nuclear translocation of phosphorylated ERKs, whereas a slower activation of ERKs (10 min) mediated by beta-arrestins resulted in the cytosolic retention of the phosphorylated ERKs. However, we observed distinct differences from this established ERKs cellular itinerary with the mu-opioid receptor-activated ERKs. Agonists such as morphine and methadone activated ERKs via the protein kinase C-dependent pathway but not the beta-arrestin-dependent pathway. The activated ERKs did not translocate into the nucleus, but phosphorylated 90-kDa ribosomal S6 kinase and induced the activity of transcription factor cAMP response element-binding protein. In contrast, agonists such as etorphine and fentanyl activated ERKs in a beta-arrestin-dependent manner. The phosphorylated ERKs translocated into the nucleus, resulting in increases in Elk-1 activity and GRK2 and beta-arrestin2 transcriptions. Thus, the cellular location of phosphorylated ERKs and subsequent activities on gene transcriptions are dictated by the agonist used to activate the receptor and the subsequent signaling pathway involved.
PMID:17947509 Full text: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253657 Zheng H et al; Mol Pharmacol 73 (1): 178-90 (2008)
It has been proposed that opioid agonist efficacy may play a role in tolerance and the regulation of opioid receptor density. To address this issue, the present studies estimated the in vivo efficacy of three opioid agonists and then examined changes in spinal mu-opioid receptor density following chronic treatment in the mouse. In addition, tolerance and regulation of the trafficking protein dynamin-2 were determined. To evaluate efficacy, the method of irreversible receptor alkylation was employed and the efficacy parameter tau estimated. Mice were injected with the irreversible mu-opioid receptor antagonist clocinnamox (0.32-25.6 mg/kg, i.p), and 24 hr later, the analgesic potency of s.c. morphine, oxycodone and etorphine were determined. Clocinnamox dose-dependently antagonized the analgesic effects of morphine, etorphine and oxycodone. The shift to the right of the dose-response curves was greater for morphine and oxycodone compared to etorphine and the highest dose of clocinnamox reduced the maximal effect of morphine and oxycodone, but not etorphine. The order of efficacy calculated from these results was etorphine>morphine>oxycodone. Other mice were infused for 7 days with oxycodone (10-150 mg/kg/day, s.c.) or etorphine (50-250 ug/kg/day, s.c.) and the analgesic potency of s.c. morphine determined. The low efficacy agonist (oxycodone) produced more tolerance than the high efficacy agonist (etorphine) at equi-effective infusion doses. In saturation binding experiments, the low efficacy opioid agonists (morphine, oxycodone) did not regulate the density of spinal mu-opioid receptors, while etorphine produced approximately 40% reduction in mu-opioid receptor density. Furthermore, etorphine increased spinal dynamin-2 abundance, while oxycodone did not produce any significant change in dynamin-2 abundance. Overall, these data indicate that high efficacy agonists produce less tolerance at equi-effective doses. Furthermore, increased efficacy was associated with mu-opioid receptor downregulation and dynamin-2 upregulation. Conversely, lower efficacy agonists produced more tolerance at equi-effective doses, but did not regulate mu-opioid receptor density or dynamin-2 abundance. Taken together, these studies indicate that agonist efficacy plays an important role in tolerance and regulation of receptors and trafficking proteins.
PMID:17349996 Full text: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1995431 Pawar M et al; Eur J Pharmacol 563 (1-3): 92-101 (2007)
We have recently reported that the antinociception induced by etorphine given i.c.v. is mediated in part by the stimulation of both mu- and epsilon-opioid receptors and the activation of both monoaminergic and opioidergic descending pain control systems. Since the opioid epsilon-receptor-mediated antinociception induced by beta-endorphin is mediated by the release of [Met]enkephalin and subsequent stimulation of delta-opioid receptors in the spinal cord, the present studies were designed to determine if beta-endorphin-like action is also involved in etorphine-induced antinociception. The tail-flick test was used to assess the antinociceptive response performed in male ICR mice. Etorphine at doses from 5 to 20 ng given i.c.v. produced a dose-dependent inhibition of the tail-flick response. The inhibition of the tail-flick response induced by etorphine given i.c.v. was antagonized by intrathecal pretreatment for 60 min with antiserum against [Met]enkephalin (10 microg), but not with antiserum against [Leu]enkephalin (10 microg) or dynorphin A (1-13) (10 microg). Desensitization of delta-opioid receptors in the spinal cord by intrathecal pretreatment with [Met]enkephalin (5 microg) for 60 min attenuated i.c.v. administered etorphine-induced tail-flick inhibition. However, intrathecal pretreatment with [Leu]enkephalin (5 microg) or dynorphin A (1-17) (0.1 microg) for 60 min did not attenuate i.c.v. administered etorphine-induced tail-flick inhibition. The results indicate that antinociception induced by etorphine given i.c.v. is mediated in part by the stimulation of the epsilon-opioid receptor at the supraspinal sites and by the release of [Met]enkephalin, which subsequently stimulates delta-opioid receptors in the spinal cord.
PMID:9284359 Xu JY, Tseng LF; Neuroscience 80 (2): 579-85 (1997)






