

1. (+)-osilodrostat
2. 4-((5r)-6,7-dihydro-5h-pyrrolo(1,2-c)imidazol-5-yl)-3-fluoro-benzonitrile
3. Benzonitrile, 4-((5r)-6,7-dihydro-5h-pyrrolo(1,2-c)imidazol-5-yl)-3-fluoro-
4. Isturisa
5. Lci699
6. Osilodrostat
1. 1315449-72-9
2. Lci699-aza
3. Unii-y6581yaw9v
4. Osilodrostat Phosphate [usan]
5. Y6581yaw9v
6. Isturisa
7. Osilodrostat(lci699) Phosphate
8. Osilodrostat (phosphate)
9. 4-((5r)-6,7-dihydro-5h-pyrrolo(1,2-c)imidazol-5-yl)-3-fluorobenzonitrile Dihydrogen Phosphate
10. 4-[(5r)-6,7-dihydro-5h-pyrrolo[1,2-c]imidazol-5-yl]-3-fluorobenzonitrile;phosphoric Acid
11. Benzonitrile, 4-((5r)-6,7-dihydro-5h-pyrrolo(1,2-c)imidazol-5-yl)-3-fluoro-, Phosphate (1:1)
12. Isturisa (tn)
13. 4-[(5r)-6,7-dihydro-5h-pyrrolo[1,2-c]imidazol-5-yl]-3-fluorobenzonitrile Dihydrogen Phosphate
14. Chembl3707393
15. Schembl13837602
16. Dtxsid401027857
17. Osilodrostat Phosphate [mi]
18. Osilodrostat Phosphate (jan/usan)
19. Osilodrostat Phosphate [jan]
20. Hy-16276a
21. Akos040749099
22. Osilodrostat Phosphate [who-dd]
23. Da-76469
24. Osilodrostat Phosphate [orange Book]
25. Cs-0068058
26. D11062
27. Q27294304
28. 4-((5r)-6,7-dihydro-5h-pyrrolo(1,2-c)imidazol-5-yl)-3-fluorobenzonitrile Monophosphate
1. Lci699
2. Osilodrostat
3. Osilodrostat (lci699)
4. Osilodrostat Free Base
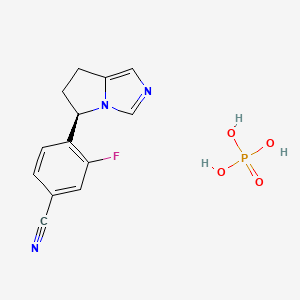
| Molecular Weight | 325.23 g/mol |
|---|---|
| Molecular Formula | C13H13FN3O4P |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 1 |
| Exact Mass | g/mol |
| Monoisotopic Mass | g/mol |
| Topological Polar Surface Area | 119 |
| Heavy Atom Count | 22 |
| Formal Charge | 0 |
| Complexity | 387 |
| Isotope Atom Count | 0 |
| Defined Atom Stereocenter Count | 1 |
| Undefined Atom Stereocenter Count | 0 |
| Defined Bond Stereocenter Count | 0 |
| Undefined Bond Stereocenter Count | 0 |
| Covalently Bonded Unit Count | 2 |
Isturisa is indicated for the treatment of endogenous Cushing's syndrome in adults.
H02CA02






