
BLOG
MARKET INTEL by PharmaCompass
CONTENT by Suppliers
- Interview #SpeakPharma
- Video #SupplierSpotlight
- Vlog #PharmaReel
- Company Bio #AboutSupplier
- Services Bio #AboutCapabilities
News
Create content with us, ask us
By PharmaCompass
2019-02-14
Impressions: 6802
The heparin adulteration scandal had engulfed the world of pharmaceuticals in 2008. Ten years on, the spotlight returned to drug manufacturing as millions of blood pressure (BP) medicines were recalled due to the risk of them containing potential cancer-causing impurities.
The ‘sartan’ recall was triggered in July 2018 when the European Medicines Agency (EMA) began reviewing medicines containing valsartan, an active pharmaceutical ingredient (API) supplied by Chinese drugmaker Zhejiang Huahai Pharmaceutical.
The company had detected the presence of an impurity N-nitrosodimethylamine (NDMA), a probable human carcinogen, in the valsartan API it supplied to several manufacturers of BP medicines available in the EU. The presence of NDMA was unexpected and is thought to be related to changes in the way the API was manufactured.
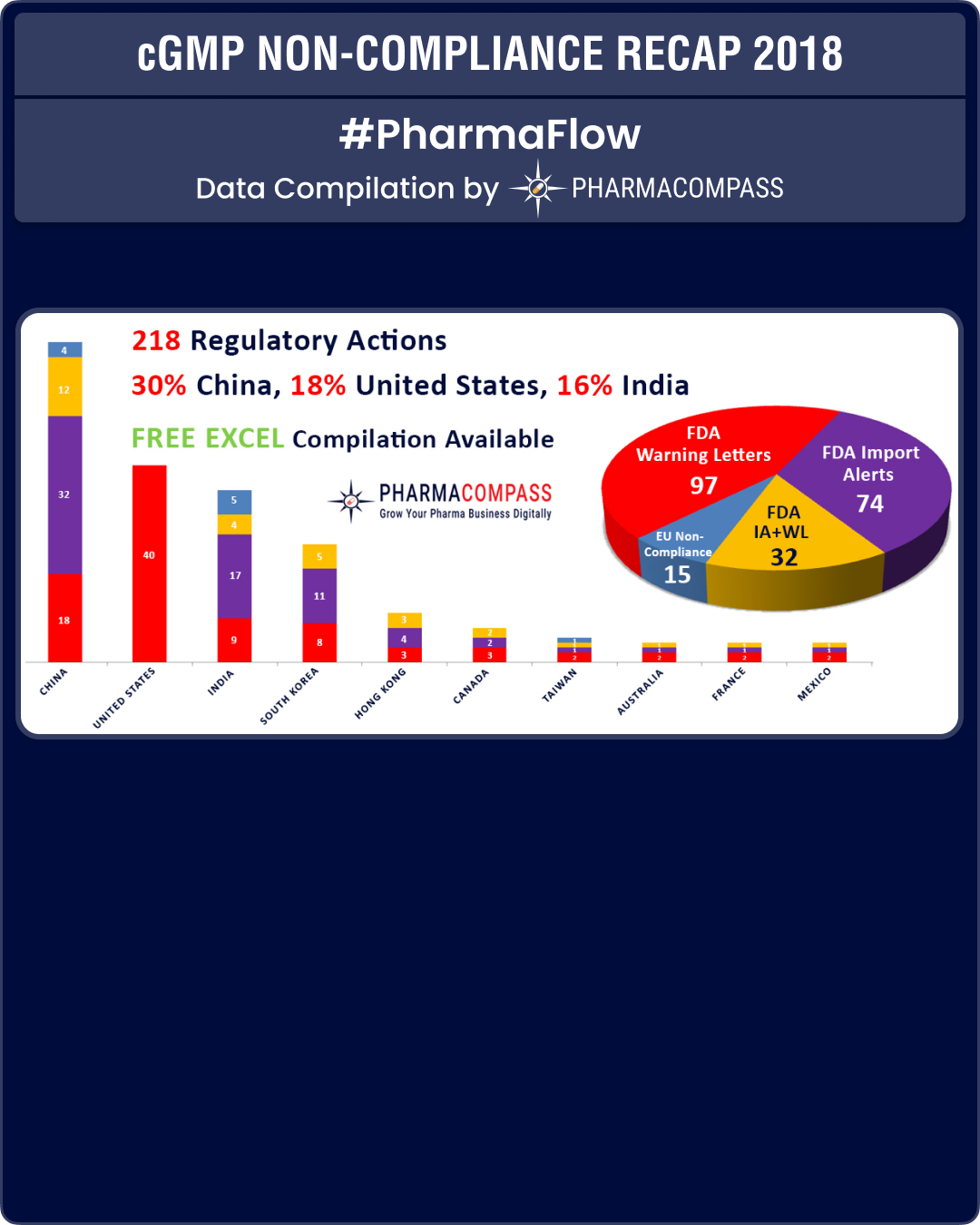
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
In our mid-year recap, we had shared the shortcomings of major pharmaceutical companies like Pfizer, Bayer and Akorn. The sartan recalls which started in the second half of 2018, highlighted the deficiencies plaguing the GMP inspection system.
The issues also brought to fore the fragility and interconnectedness of the global pharmaceutical supply chain, along with concerns over lack of assurance on the quality of the marketed drug products.
Deficiencies in
the GMP inspection system
In 2007, Huahai became the first Chinese drugmaker to receive FDA approval for a finished drug product (nevirapine). Even today, it ranks amongst the top Chinese drug master file (DMF) filers with the FDA.


Following the concerns over its valsartan API, the FDA issued a warning letter to the firm after the agency conducted a two-week inspection at Huahai’s API manufacturing facility at Linhai (Zhejiang province, China) in the last week of July through the first week of August.
The inspection revealed that the manufacturing process change, which most likely resulted in the generation of the NDMA impurity, was approved in 2011. This would mean that the tainted ingredient had been on the market for years, not weeks or months.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
The 11-observation, highly redacted Form 483 issued to Huahai after the 2018 inspection also revealed the absence of a formal risk assessment of critical changes made which could impact the quality of intermediates or APIs produced at the site.
In December 2013, Huahai had filed an amendment to its Drug Master File, indicating that the amendment was submitted for minor changes for drug substance manufacturing, when its own internal change classified the change as a critical one.
Huahai was also found to release finished APIs manufactured from crude intermediates with OOS (out of specifications) levels of genotoxic impurities without conducting a thorough investigation.
Between December 2016 and August 2017, Huahai initiated 17 OOS investigations for failure of an impurity specification (potentially genotoxic) and in all 17 cases, the batches were reprocessed without developing a preventive action plan as 13 out of the 17 OOS results were attributed to lab error, five to production errors and two were believed to be a combination of lab and production errors.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
Since the sartan manufacturing process change was implemented in 2011, an effective FDA inspection should have raised concerns in 2014. Instead, it found no observations whatsoever. In 2017, when their own investigator mentioned that the firm was not ready for API manufacturing and raised concerns over data integrity and their handling of OOS issues, the FDA still chose not to act as per the recommendations of the investigator.
Unsurprisingly, following the recalls, a September 2018 inspection by investigators from the Italian Ministry of Health raised data-integrity concerns in relation to GC-FID (gas chromatograph-flame ionization detector) analysis and found that the company conducted inadequate investigation of unknown peaks detected in GC-MS (gas chromatography–mass spectrometry) analysis of batches of valsartan manufactured with the new process.
The Mylan warning letter that was never sent
The issue of concerns at Huahai not being flagged in time is not an isolated one. Bloomberg recently carried a series of news features on the risks facing America’s generic drug industry.
One of the articles highlighted an inspection at Mylan where inspectors found bins full of shredded documents, including quality-control records. According to the article, “a warning letter, the FDA’s strongest rebuke, was drafted. It would mean the agency could refuse to consider any Mylan application for a new drug made at that plant until the company fixed things.”
“But the warning letter was never sent. Eight months later, in July 2017, higher ups at the FDA apparently decided to take it easy on Mylan, the second-largest generic drugmaker, with US$ 12 billion in revenue in 2017. The FDA ultimately left it in the company’s hands, and the drugmaker promised to address what the agency calls a ‘data integrity’ issue.”
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
In April 2018, the FDA completed an inspection of Mylan’s manufacturing site in Morgantown and the Form 483 highlighted serious quality concerns ranging from the lack of oversight by the quality unit to cross contamination concerns.
According to a list of laboratory investigations generated during the inspection, 4,279 out of 25,432 investigations were not closed by quality assurance (QA) prior to batch release. Of these open investigations, 1,945 investigations were for in-process results that were out of specification.
The FDA issued a warning letter on November 9, 2018.
Fragility
of the global pharmaceutical supply chain
Had the valsartan issue been a standalone one, it would have been cast off as an issue plaguing only one drug manufacturer. However, the issue spread like wildfire as the review was subsequently extended to other manufacturers and other ‘sartan’ medicines.
Immediately after the Huahai alert, two Chinese companies — Zhejiang Tianyu Pharmaceutical and Zhuhai Rundu Pharmaceutical — stepped forward to say they were recalling BP drugs tainted with NDMA.
The EMA announced that the valsartan API manufactured by Zhejiang Tianyu Pharmaceutical had NDMA detected in it, though the levels were much lower as compared to what was seen in the API from Zhejiang Huahai.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
However, the EMA said Zhejiang Tianyu was no longer authorized to manufacture the valsartan API for EU , following the suspension of its CEP (a certificate of suitability verifying that the quality of its valsartan meets European requirements).
Post EMA action against the Chinese manufacturer, a unit of India’s Hetero Drugs joined the group of companies that announced valsartan recalls in the US.
The notice from the FDA said Hetero was found to be using a similar manufacturing process as China’s Zhejiang Huahai for valsartan.
Major generic players like Mylan and Teva Pharmaceuticals also began recalling valsartan products after Mylan reported that another cancer-causing impurity — N-Nitrosodiethylamine (NDEA) — was found in some of the API made at one of its India plants and Teva had used that API to manufacture its products.
As the concern expanded to other ‘sartan’ medicines, EMA found very low levels of NDEA in the losartan made by Hetero Labs in India.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
This discovery resulted in Sandoz announcing a recall of losartan that was manufactured by its Lek Pharmaceuticals unit in Ljubljana, Slovenia. Sandoz also issued a voluntary recall of one lot of losartan potassium and hydrochlorothiazide and then there was news that Torrent Pharmaceuticals Limited also voluntarily recalled two lots of losartan potassium tablets.
After losartan, irbesartan became the third sartan to be found with low levels of NDEA. On October 8, 2018, the European Directorate for the Quality of Medicines & HealthCare (EDQM) suspended Aurobindo Pharma’s CEP, effectively stopping the supply in the EU of medicines containing irbesartan from this company.
Alerts and recalls were undertaken by all major regulatory authorities in countries such as France, the UK, Portugal, Germany, Finland, Ireland, Bahrain, Bulgaria, Italy and Taiwan.
Lack
of assurance on product quality
Six months on, despite the FDA following an exhaustive effort, in collaboration with global regulators and a multidisciplinary team of experts, the agency is yet to know the root cause of the nitrosamine impurities found in the sartans. Investigations are still on.
According to the FDA, the probable cancer-causing impurities “may be generated when specific chemicals and reaction conditions are present in the manufacturing process of the drug’s API, and may also result from the reuse of materials, such as solvents.”
Given the fact that the exact root cause of these impurities is still not known, testing for the presence of these impurities is the only safety measure currently being suggested by regulators.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
For this reason, the EMA has said “companies must now take measures to avoid the presence of these impurities and carry out rigorous testing of their products.”
In the wake of the sartan scandal, one wonders if other products on the market also have unknown impurities, which are not being tested for (as was the case of NDMA and NDEA in the past).
Given such a scenario, how does one know when the next sartan-like problem may crop up?
The vaccine
scandal in China
It was not just the sartan scandal that rocked the world of pharmaceuticals. In Northeast China, a vaccine producer in Jilin province was forced to pay about 9.1 billion yuan (US$ 1.3 billion) in penalties for law violations in producing rabies vaccines for human use.
Changchun Changsheng Bio-tech Co, in Changchun, was found to have serious irregularities, including fabricating production records in the manufacture of rabies vaccines for human use, during an inspection by the State Drug Administration, China FDA said in a statement.
Changsheng's illegal activities included using expired vaccine raw materials, altering production dates, forging permits and destroying evidence during inspections.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
The Changsheng case revealed many loopholes such as the lack of supervision and oversight by the former State Food and Drug Administration and the State Drug Administration.
China said it has sacked six senior officials at its food and drug regulator after a safety scandal at vaccine maker Changsheng Biotechnology Co. Ltd revealed failings at the government body, including inadequate supervision.
Our view
As was the case in 2017, in 2018 too China, India and the US continued to be the top three countries where regulators uncovered compliance issues.
With South Korea emerging as a major hub for drug manufacturing, there was a lot more FDA action against companies in the country.
While data-integrity violations and a failure to thoroughly investigate deviations continued to remain a major concern for inspectors, the real concern emanated from the supply of product to market (which had the potential to impact product quality or patient safety).
On the one hand, FDA placed the Huahai facility making valsartan on import alert to stop all its API and finished drugs that use Huahai’s API from legally entering the US, on the other hand the Chinese city government of Linhai (in the eastern province of Zhejiang, where the company is based) gave the company 300 million yuan (US$ 43 million).
Huahai described the payout as an “industrial development assistance fund”. Therefore, in order to overcome these regulatory concerns, greater alignment is necessary among regulators across the world.
View our Interactive cGMP Non-Compliance Recap 2018 Dashboard (Free Excel Available)
The PharmaCompass Newsletter – Sign Up, Stay Ahead
Feedback, help us to improve. Click here
Image Credit : cGMP Non-Compliance Recap 2018 by PharmaCompass is licensed under CC BY 2.0
“ The article is based on the information available in public and which the author believes to be true. The author is not disseminating any information, which the author believes or knows, is confidential or in conflict with the privacy of any person. The views expressed or information supplied through this article is mere opinion and observation of the author. The author does not intend to defame, insult or, cause loss or damage to anyone, in any manner, through this article.”



