BLOG
MARKET INTEL by PharmaCompass
CONTENT by Suppliers
- Interview #SpeakPharma
- Video #SupplierSpotlight
- Vlog #PharmaReel
- Company Bio #AboutSupplier
- Services Bio #AboutCapabilities
News
Create content with us, ask us
By PharmaCompass
2021-02-18
Impressions: 4039
The coronavirus pandemic changed the way we lived our lives, and regulatory agencies were no exception to that rule. In early March 2020, the US Food and Drug Administration (FDA) announced that it was postponing most inspections outside the United States. And the agency said only foreign inspections “deemed mission-critical” will be considered on a case-by-case basis.
As an interim measure, the FDA ensured the safety of products imported into the US through physical examinations and/or product sampling at borders, by reviewing a firm’s compliance history and by requesting records “in advance of or in lieu of” on-site drug inspections.
By the end of 2020, FDA and the European Directorate for the Quality of Medicines & HealthCare (EDQM) had managed to complete over 2,200 inspections across the world. According to a report published on the Regulatory Affairs Professionals Society (RAPS) website, across all approval types, the number of cases where pre-approval inspections by the FDA were skipped in favor of alternative approaches rose from 48 percent in July-September 2020 to 60 percent by October-December 2020.
Although inspections were down in 2020, the FDA’s Center for Drug Evaluation and Research (CDER) met its user fee commitments.
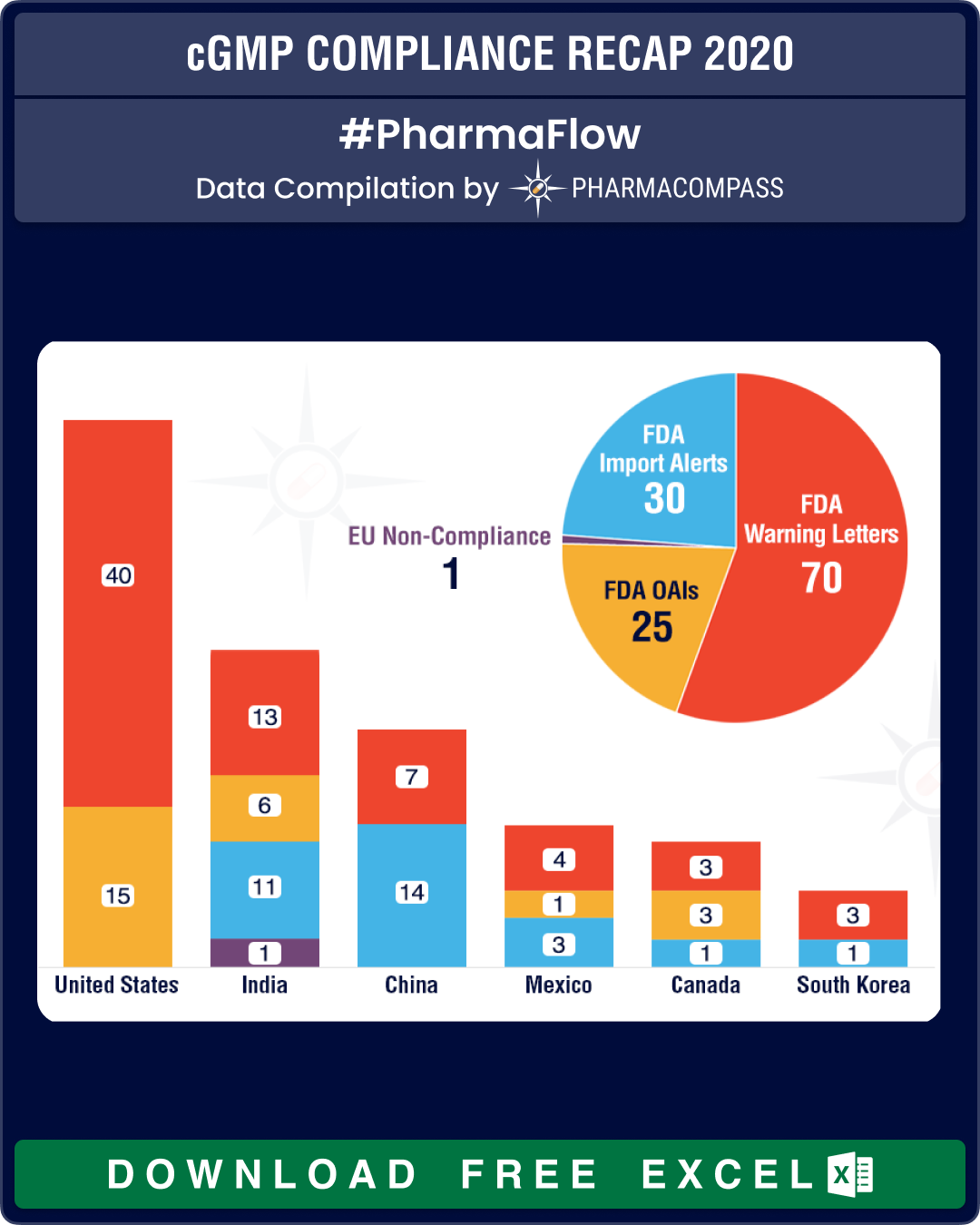
The FDA was the most active locally and issued almost 40 warning letters to drug manufacturers based in the US. Consistent with the previous years’ trend, warning letters in the US were followed by those issued to manufacturers in India (13) and China (7).
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Celgene investors lose out on deal sweetener due to FDA inspection delays
The suspension of inspections impacted Bristol Myers Squibb the most. It was hopeful of getting an FDA approval for its CAR-T therapy — lisocabtagene maraleucel (liso-cel) — for the treatment of adults with relapsed or refractory large B-cell lymphoma after at least two prior therapies. However, the FDA extended the action data by three months due to the pandemic, with no decision taken even after its November 2020 deadline.


The FDA approval of liso-cel by December 31, 2020 was one of the milestones of the contingent value rights (CVRs) issued upon the close of the Celgene acquisition by Bristol Myers to its investors. Since the approval never came through by December-end, Celgene investors lost out on the deal sweetener.
One of the reasons for the delayed approval was the inspection of a Lonza facility (which is under contract to make viral vectors for liso-cel) which was finally inspected in December 2020. The Form 483 issued last month revealed problems such as lack of storage area controls and inadequate measures to prevent microbial contamination. Liso-cel finally bagged the FDA nod this month.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Manufacturing problems, data-integrity issues drive Akorn to bankruptcy
Akorn, which has seen a series of problems over the last few years, filed its Chapter 11 petition in the Delaware bankruptcy court last year. The Illinois-based company listed as much as US$ 10 billion in debt and US$ 10 billion in assets in its Chapter 11 petition.
The drugmaker said it had given up on finding itself a buyer amidst the uncertainties brought about by the pandemic.
In April 2018, Fresenius had walked away from a US$ 4 billion takeover of Akorn, citing a breach of FDA’s data integrity requirements by Akorn. Its shareholders then sued Akorn for concealing data integrity problems. In August 2019, Akorn settled the matter with its shareholders.
Akorn has received several FDA warning letters over the last few years, including those issued to its Somerset (New Jersey) facility in June 2019, to its Decatur (Illinois) facility in January 2019 and to its Amityville (New York) facility in February 2019.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Ipca placed back on FDA import alert after US overcomes chloroquine shortage
In March 2020, when chloroquine (CQ) and hydroxychloroquine (HCQ) were considered possible treatments for Covid-19, the FDA had partially lifted an import alert on Ipca’s two plants to ensure the supply of chloroquine tablets. The exemption had allowed Ipca to export APls chloroquine phosphate and hydroxychloroquine sulfate from the company’s API manufacturing facility situated in Ratlam (Madhya Pradesh, India).
In June, once questions were raised around the efficacy of chloroquine in treating Covid, FDA said no shipment of API chloroquine phosphate will be excluded from the import alert.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Data-integrity concerns at Pfizer’s India unit; warning letters to Mylan
In 2019, when the mega-merger of Mylan with Upjohn (Pfizer’s off-patent branded and generic established medicines business) was announced, PharmaCompass had highlighted how compliance would play a key role in determining the merger’s success.
Early last year, Pfizer’s sterile manufacturing operations in India received a warning letter from the FDA. The site, located in Visakhapatnam (Andhra Pradesh, India), was inspected from August 29 to September 6, 2019.
The warning letter highlighted that Pfizer’s operations failed to conduct adequate investigations, including the timely implementation of effective corrective action and preventive action (CAPA) plans.
The Visakhapatnam operation was acquired by Pfizer in February 2015 as part of its US$ 17 billion acquisition of Hospira. When the deal was struck, Pfizer was aware of Hospira’s manufacturing record as the company was issued FDA warning letters in four out of seven continents.
Last year, the FDA also issued a warning letter to Mylan's API
manufacturing facility in India
which had issues related to enhanced cross-contamination risk and inadequate
cleaning procedures.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Serious cross-contamination problems at Cipla; data-integrity issues at Windlas
Indian drugmaker Cipla’s finished pharmaceuticals manufacturing facility in Goa received a 38-page Form 483 following a September 2019 inspection, and the warning letter issued to the facility highlighted severe cross contamination concerns.
During the inspection, FDA investigators observed residue on manufacturing equipment which when tested by Cipla confirmed the presence of multiple APIs. In its warning letter, the agency states that there is no assurance that the scope of Cipla’s evaluation into this cross-contamination problem was comprehensive.
Following the inspection, Cipla informed the FDA of its decision to suspend production in its sterile units where the FDA had raised concerns over atypical High Efficiency Particulate Air (HEPA) filter failures.
The FDA also issued a warning letter to Indian finished formulation manufacturer Windlas Healthcare, which was placed on an import alert by the FDA on January 21, 2020. Indian generic drugmakers Panacea Biotech and Shilpa Medicare were also issued FDA warning letters in 2020.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Apotex’s API unit in Mexico receives FDA warning letter
The manufacturing operations of Canadian generic drug maker, Apotex, continued to run into problems with the FDA as its API manufacturing facility in Mexico — Signa SA de CV — received a warning letter following an inspection from December 16 to 20, 2019. As in previous cases, this warning letter too highlighted cGMP deviations.
The key concern highlighted by the FDA was that the facility undertook inadequate investigations into out-of-specification (OOS) test results. The agency said Signa failed to appropriately justify potential root causes, expand investigations to all potentially affected batches, and implement adequate CAPA.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
Our view
Although the FDA had halted all physical inspections last year due to the pandemic, last month the agency announced the resumption of inspections in China and India in a bid to keep drug approvals on schedule.
On January 27, 2021, Patrizia Cavazzoni, the acting director of CDER, said in a tweet: “FDA has begun conducting prioritized inspections by investigator staff in China and is planning to initiate prioritized inspections in India shortly.”
Earlier this month, Alembic Pharmaceuticals said the FDA has made five observations post an inspection of its new injectable facility at Karkhadi (Gujarat, India) from January 29 to February 5.
With the restart of inspections in Asia, it’s only a matter of time before we see a significant increase in regulatory actions by the regulators.
View cGMP Non-Compliance Recap 2020 Dashboard (Free Excel Available)
The PharmaCompass Newsletter – Sign Up, Stay Ahead
Feedback, help us to improve. Click here
Image Credit : cGMP Compliance Recap 2020 by PharmaCompass is licensed under CC BY 2.0
“ The article is based on the information available in public and which the author believes to be true. The author is not disseminating any information, which the author believes or knows, is confidential or in conflict with the privacy of any person. The views expressed or information supplied through this article is mere opinion and observation of the author. The author does not intend to defame, insult or, cause loss or damage to anyone, in any manner, through this article.”